Enhancements to plot_segments function? #34
Comments
|
Hi @sigven, thanks for your comments:
|
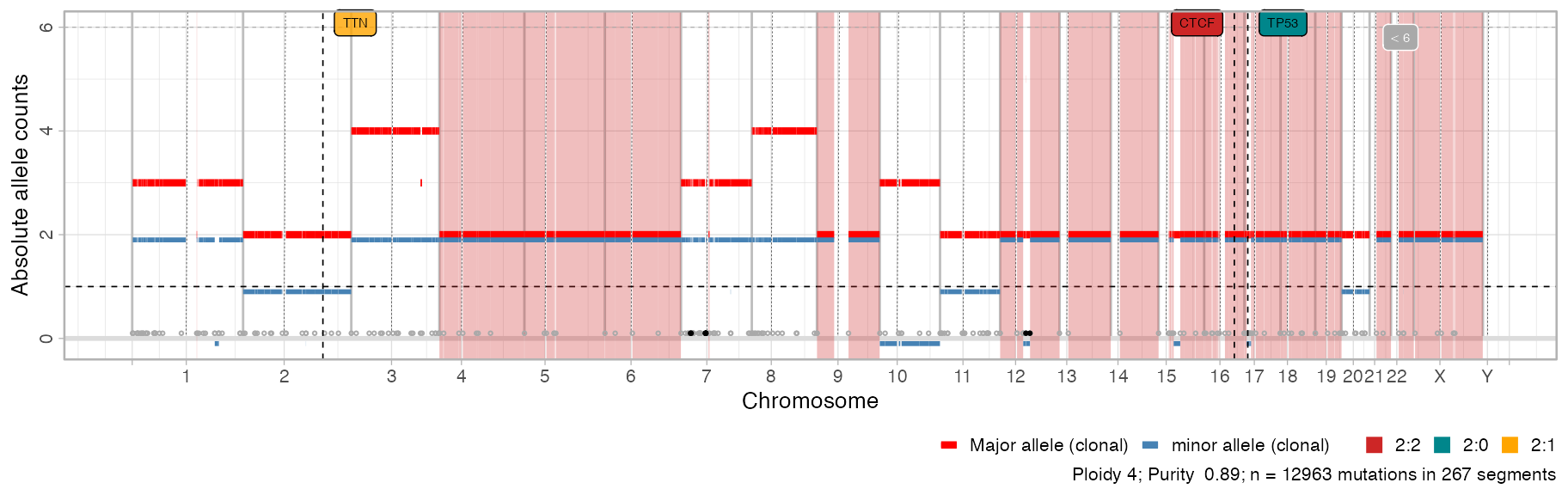{kind=link}
|
Hi @sigven still interested on this? |
|
Hi @caravagn, yeah indeed! Sorry for not responding, I hope to get back to you shortly. |
|
Hi @caravagn (cc @pdiakumis), Truly sorry for the very late response and follow-up on this matter. We have been working hard to use your package inside PCGR, where we want to plot allele-specific copy number input from tumor samples for clinical interpretation. Notably, PCGR relies upon the Conda framework, and we have struggled to make kind regards, |
|
Hi @sigven this is more clear now; I would love to see it used in your tool. Maybe we can help with this, let me talk with the guys and see if we can consider improving dependencies (some might be not strictly required). Could you please clarify in which form you would like the package to be installable? Is the problem that the package depends on too many other packages for your internal use? @luca-dex I might need you help on this. |
|
@luca-dex Regarding dependencies I think that
|
|
Hello Giulio, |
|
Hi @pdiakumis, I see, thanks for clarifying, I can then close this. Then maybe the best for you is to re-use our code from
Best, Giulio |
Hi,
Thanks for a very nice package with lots of great functionality for copy-number processing and visualization. This is really missing from other packages. Great work! I have been playing a bit with data from our tumor samples, and want to share some thoughts on the visualization function
plot_segments:plot_segmentsto plotly, i.e.plotly::ggplotly(CNAqc::plot_segments(x)), which, if working optimally, would give the users a powerful oppertunity to interact with the segments (and potentially annotations therein), being able to zoom in on particular chromosomes etc. I thought that restricting thechromosomesargument inplot_segmentswould give me a higher-resolution view of a particular chromosome, but it seems that the whole genome track is also plotted for this?Happy to get your input on these matters:-)
kind regards,
Sigve
The text was updated successfully, but these errors were encountered: