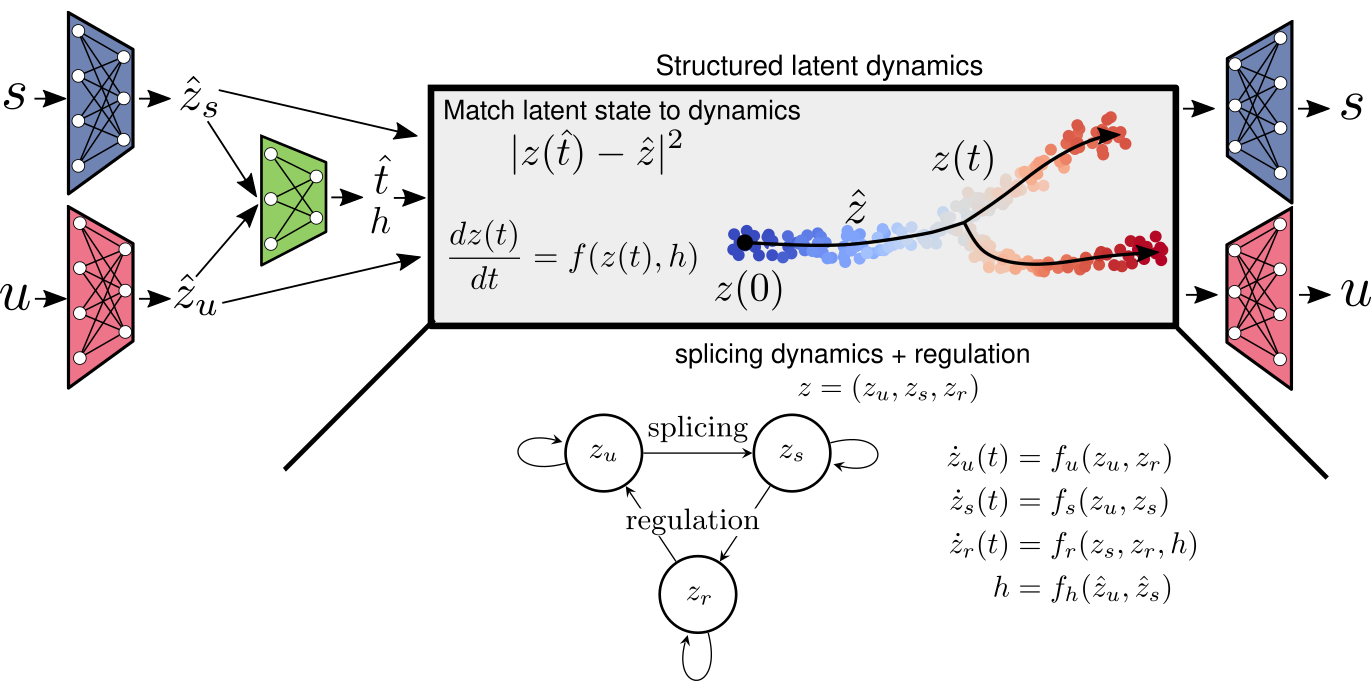
Estimating RNA velocity in a learned latent space, enabling batch correction and dynamics based embeddings.
Pre-print available at https://www.biorxiv.org/content/10.1101/2022.08.22.504858v2
The paper_notebooks/ directory runs LatentVelo on all of the datasets used in the paper. The examples/ directory shows a documented example of LatentVelo on synthetic data.
Benchmarking plots are generated by the notebooks in the benchmarks/ directory. Subdirectories of this directory contain code to run benchmarking with synthetic data or batch correction. Additionally the code used to run scVelo is here as well.
Additional settings are available in DOCUMENTATION.md. Information about acquiring the datasets used is in DATASETS.md
Currently not avaiable on pip, just download the repo and install with
python setup.py install
in the main directory.
LatentVelo uses AnnData annotated data objects. This object must have two layers containing spliced and unspliced counts.
Data is prepared for use with LatentVelo as follows:
ltv.utils.standard_clean_recipe(adata, spliced_key='spliced', unspliced_key='unspliced',
batch_key='batch', celltype_key='celltype')
Batch and celltype keys for the standard model are optional. For the celltype-annotated model, the following function is used to prepare data and must include a celltype key:
ltv.utils.anvi_clean_recipe(adata, spliced_key='spliced', unspliced_key='unspliced',
batch_key='batch', celltype_key='celltype')
The LatentVelo model can be initialized as a standard VAE or a celltype annotated VAE:
model = ltv.models.VAE(observed = number_of_genes, latent_dim = latent_dimension,
zr_dim = latent_regulation_dimension,
h_dim = conditioning_dimension)
model = ltv.models.AnnotVAE(observed = number_of_genes, latent_dim = latent_dimension,
zr_dim = latent_regulation_dimension,
h_dim = conditioning_dimension,
celltypes = number_of_celltypes)
Batch correction is enabled by specifying batch correction and the number of batches for either model:
model = ltv.models.VAE(observed = number_of_genes, latent_dim = latent_dimension,
zr_dim = latent_regulation_dimension,
h_dim = conditioning_dimension,
batch_correction = True,
batches = number_of_batches)
The model is trained with the following function, and validation set autoencoder and trakectory reconstruction losses are output:
epochs, val_ae, val_traj = ltv.train(model, adata, batch_size = batch_size,
epochs=number_of_epochs,
name=parameter_output_folder_name)
The following function is used to output the results of LatentVelo to a new AnnData object containing the results on the LatentVelo latent space. If desired, gene velocities can also be included. Model reconstructions using the decoder for both the autoencoder and trajectories can also be included.
latent_adata, adata = ltv.output_results(model, adata,
gene_velocity = True,
decoded = True,
embedding='umap')
scVelo can then be used to plot 2D velocity streamlines:
scv.tl.velocity_graph(latent_adata, vkey='spliced_velocity')
scv.pl.velocity_embedding_stream(latent_adata, vkey='spliced_velocity',
color='latent_time')
To output cell trajectories:
z_traj, times = ltv.cell_trajectories(model, adata)
These can then be plotted on the latent space UMAP plot.
LatentVelo was run with the packages
torchdiffeq 0.2.2
pytorch 1.11.0
seaborn 0.11.2
scvi-tools 0.15.0
scvelo 0.2.4
scipy 1.8.1
sklearn 1.1.1
scanpy 1.9.1
scgen 2.1.0
pandas 1.4.2
numpy 1.22.4
anndata 0.8.0
unitvelo 0.1.5
scib 1.0.3
matplotlib 3.5.2
If you find this useful please cite
@article {Farrell2022.08.22.504858,
author = {Farrell, Spencer and Mani, Madhav and Goyal, Sidhartha},
title = {Inferring single-cell transcriptomic dynamics with structured latent gene expression dynamics},
elocation-id = {2022.08.22.504858},
year = {2022},
doi = {10.1101/2022.08.22.504858},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2022/12/01/2022.08.22.504858},
eprint = {https://www.biorxiv.org/content/early/2022/12/01/2022.08.22.504858.full.pdf},
journal = {bioRxiv}
}