![]()

A basic and quick workflow for differential expression analysis.
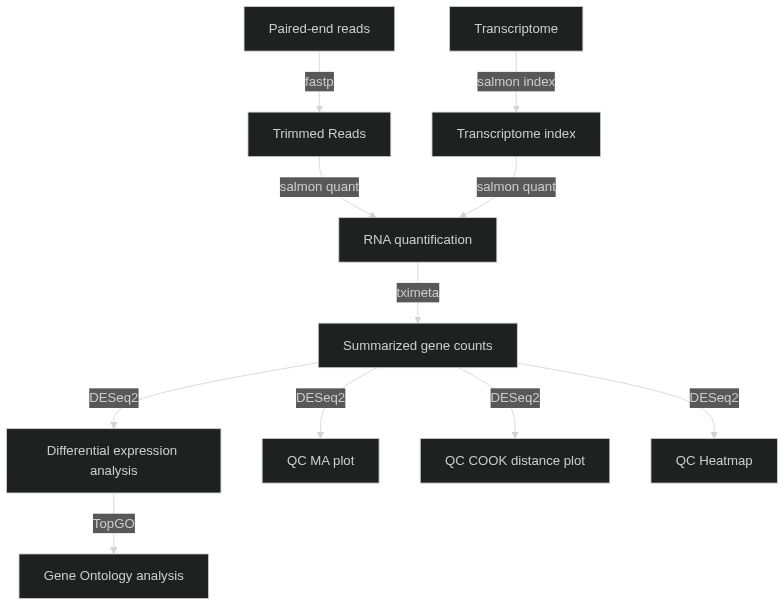
codename: mnemonic codename for the run (default: 'quick-rnaseq')outdir: directory where to store the results (default: './results')experiment.samplesheet: CSV file describing samples and conditions (required).experiment.contrasts: contrasts for differential expression analysis in the format [['case1', 'control'],['case2','control']], which performs the analysis of case1 vs control samples, and case2 vs control samples. (required)transcriptome.reference: transcriptome reference file. Gencode recommended (required. GZ format)transcriptome.decoys: reference genome file. Gencode recommended (required. GZ format)fastp.args: options for reads trimming using fastp. (default: '')salmon.index.args: options for Salmon index, e.g. '--gencode' for Gencode transcritomes.salmon.quant.libtype: library type for quantification (default: 'A', Salmon infers lib type)salmon.quant.args: Salmon options. (default: '--validateMappings --gcBias')summarize_to_gene.counts_from_abundance: infer counts from abundances using tximeta (default: 'no')summarize_to_gene.organism_db: Bioconductor organism package for annotation. Currently supportingorg.Hs.eg.dbfor Human andorg.Mm.eg.dbfor mouse and (default: 'org.Hs.eg.db')qc.pca.transform: counts transformation for PCA analysis (see DESeq2. default: 'rlog')qc.ma.lfc_threshold: log fold-change threshold for MA plot (see DESeq2. default: 0)qc.sample.transform: counts transformation for PCA analysis (see DESeq2. default: 'rlog')dge.lfc_threshold: log fold-change threshold for differential expression analysis (see DESeq2. default: 0)dge.fdr: false discovery rate threshold to be used with implicit filtering (see DESeq2. default: 0.05)gene_ontology.organism_db: Bioconductor organism package for annotation. Currently supportingorg.Hs.eg.dbfor Human andorg.Mm.eg.dbfor mouse and (default: 'org.Hs.eg.db')gene_ontology.gene_id: type of gene id used for the analysis (default: 'ensembl')gene_ontology.remove_gencode_version: remove Gencode version from gene id (default: 'yes')gene_ontology.fdr: false discovery rate threshold (default: 0.05)
nextflow pull stracquadaniolab/quick-rnaseq-nfnextflow run stracquadaniolab/quick-rnaseq-nf -profile test,dockernextflow run stracquadaniolab/quick-rnaseq-nf -profile test,singularitynextflow run stracquadaniolab/quick-rnaseq-nf -profile test,singularity,slurmPrepare a samplesheet.csv file as follows:
sample,read1,read2,condition
6C_REP1,data/RF01_6C1_R1_001.fastq.gz,data/RF01_6C1_R2_001.fastq.gz,control
6C_REP2,data/RF01_6C2_R1_001.fastq.gz,data/RF01_6C2_R2_001.fastq.gz,case
Please note that the header is required and it is case sensitive.
Prepare a nextflow.config file as follows:
params {
// experiment information
experiment.samplesheet = "./samplesheet.csv"
experiment.contrasts = [['case1', 'control'],['case2','control']]
// transcriptome information
transcriptome.reference = "gencode.v40.transcripts.fa.gz"
transcriptome.decoys = "GRCh38.primary_assembly.genome.fa.gz"
}
Now you can run quick-rnaseq as follows:
nextflow run stracquadaniolab/quick-rnaseq-nf -profile singularity,slurmresults/analysis/dge-<contrasts>.csv: file with the differential expression analysis results for a given contrast.results/analysis/go-<contrasts>.csv: file with the GO analysis results for a given contrast.results/dataset/summarized-experiment.rds: DESeqDataset object with all experimental information (e.g. gene counts)results/qc: quality control reportresults/quantification/<sample-name>: Salmon quantification folders for each sample.
- Giovanni Stracquadanio, giovanni.stracquadanio@ed.ac.uk