Home
Pindel is a pattern growth approach which can detect breakpoints of large deletions, medium-sized insertions, inversions,tandem duplications from paired-end short reads. Next we describe how you can use Pindel.
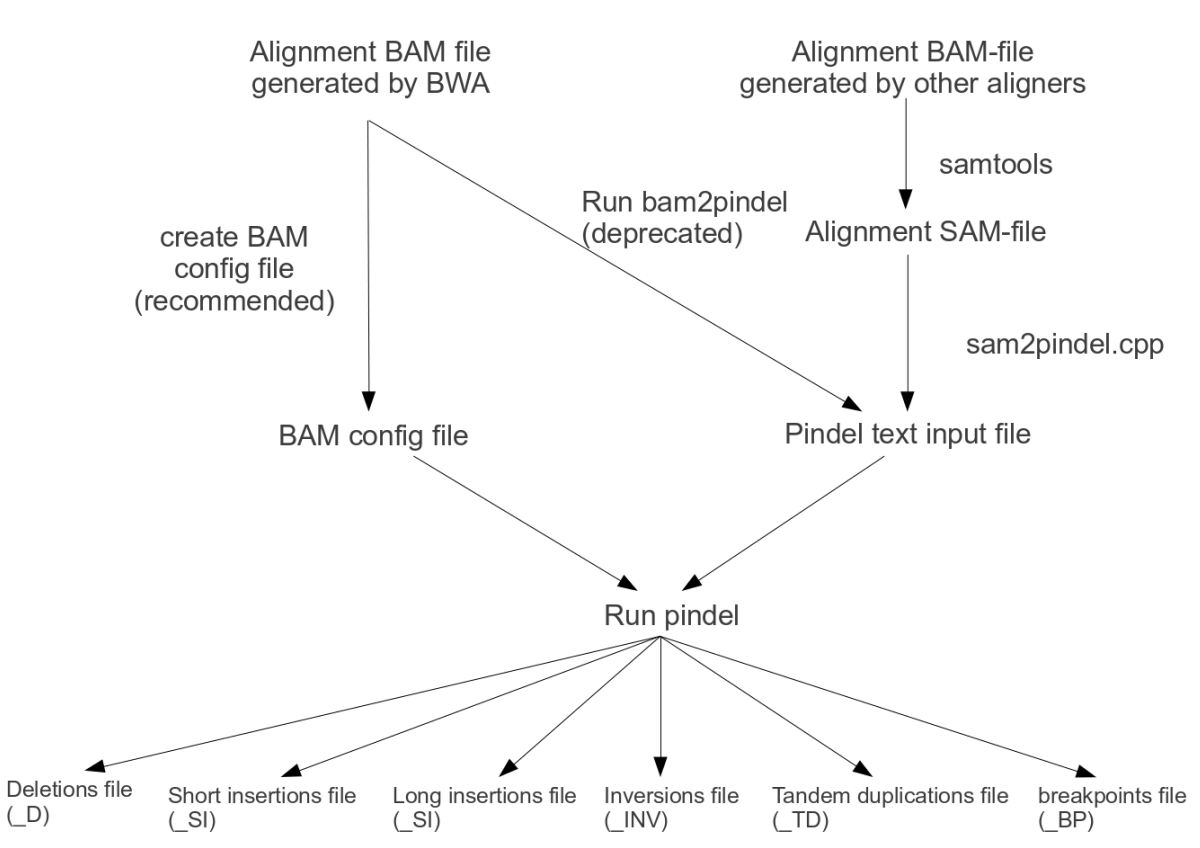
Option 1 and the best to try first: Indexed BAM files produced by BWA on Illumina reads, via a configuration file
Pindel version 0.2.0 and above can directly read in BWA generated BAM-files. As a user, you do need to create a small text-file containing the path ,the insert size(the length of sequence between the paired-end adapters in paired-end sequence),and the label of the BAM-files you want to search for indels and structural variations.
For example, given two bam-files named ‘sample_1.bam’ and ‘sample_2.bam’ with insert-sizes of 400 bp, the configuration text file could look like:
/path/to/sample_1/sample_1.bam 400 sample1
/path/to/sample_2/sample_2.bam 400 sample2
When running Pindel, use the -i option followed by the name of the config file (such as ‘config_1&2.txt’)
If another aligner than BWA was used, the best way to convert the reads to pindel input format is to use sam2pindel.cpp. Sam2pindel expects a mapping location of mate read if the query is unmapped while the mate has been mapped. Users may need to fix the information about mate reads for some mappers such as bowtie.
Compiling and running sam2pindel.cpp works as follows:
- Download the cpp code (SAM_2_PINDEL_cin_2010Dec2.cpp) from either of the following sources:
https://trac.nbic.nl/pindel/downloads
http://www.ebi.ac.uk/~kye/pindel/v_0.2.0/SAM_2_PINDEL_cin_2010Dec2.cpp
ftp://ftp.sanger.ac.uk/pub/zn1/pindel/Pindel_0.2.0.tar.gz
- Compile the cpp source code g++ SAM_2_PINDEL_cin_2010Dec2.cpp –o sam2pindel –O3
- Run sam2pindel
If the input is a sam file, the command should look similar to this:
./sam2pindel input.sam Output4Pindel.txt 400 sample1 0
If the input is a bam-file, sam2pindel should be run like this:
samtools view input.bam | ./sam2pindel - Output4Pindel.txt 400 sample2 0
Sam2pindel requires five arguments:
- Input sam file ( ‘input.sam’)
- Output for pindel ( ‘Output4Pindel’)
- Insert size (400)
- Sample tag: (‘sample1’)
- Number of extra lines (non-record and not starting with @) in the beginning of the file to skip (0)
git clone git://github.com/xjtu-omics/pindel.git
cd pindel
./INSTALL /path/to/samtools_FOLDER/
If you have a BWA-BAM-file and created a bam-configuration file as option 1, run pindel with the -i option:
./pindel -f ref.fa -i sample1.bam.config.txt -c ALL -o /path/to/store/output/of/pindel/for/sample1/
If used option 2, run pindel with the -p option followed by the name of the pindel input file:
./pindel -f ref.fa -p sample2.Output4Pindel.txt -c ALL -o /path/to/store/output/of/pindel/for/sample2/
The output files should then contain all detected indels and SVs relative to the reference.
There are some required parameters:
-f/--fasta the reference genome sequences in fasta format
-p/--pindel-file the Pindel input file (either this or a bam configuration file is required)
-i/--config-file the bam config file;(either this or a pindel input file is required)
-o/--output-prefix dir to store output files of pindel
-c/--chromosome which chr/fragment. Pindel will process reads for one chromosome each time. ChrName must be the same as in reference sequence and in read file. '-c ALL' will make Pindel loop over all chromosomes. We can limit process to a specific region by -c chr20:5,000,000-15,000,000 (report indels and SVs in the range [5M, 15M]) or -c chr20:10,000,000 (look for indels and SVs in the range [10M, end])
Parameters affecting runtime and memory usage:
-T/--number_of_threads the number of threads Pindel will use (default 1). More threads assures lower runtime, but requires multiple processors
-w/--window_size for saving RAM, divides the reference in bins of X million bases and analyzes the reads per bin(default 10 million). A smaller bin size will reduce memory but will increase runtime slightly.
-x/--max_range_index the number of searching region quadrupling of pattern growth.It will influence the maximum size of structural variations we want to detected; the higher this number, the greater the maximum size of the reported SVs, but the computational cost and memory requirements increase, as does the rate of false positives. 1=128, 2=512, 3=2,048, 4=8,092, 5=32,368, 6=129,472, 7=517,888, 8=2,071,552, 9=8,286,208 (maximum 9, default 5)
Parameters affecting SV types and other information are reported:
-r/--report_inversions report inversions (default true)
-t/--report_duplications report tandem duplications (default true)
-l/--report_long_insertions report insertions of which the full sequence cannot be deduced because of their length (default true)
-k/--report_breakpoints report breakpoints (default true)
-s/--report_close_mapped_reads report reads of which only one end (the one closest to the mapped read of the paired-end read) could be mapped (default false)
Parameters related with SV detected length are reported:
-n/--min_NT_size only report inserted (NT) sequences in deletions greater than this size (default 50)
-v/--min_inversion_size only report inversions greater than this number of bases (default 50)
--min_svlen_detect only report tandem duplications greater than this number of bases (default 50)
--max_svlen_detect only report tandem duplications smaller than this number of bases (default 100000)
There still have other parameters,please use the -h/--help command line.
Pindel raw output include several files,_D is the deletion variants reported by Pindel,_SI is short insertion,_INV is inversion,_TD is tandem duplication,_LI is large insertion,_BP is unassigned breakpoints.
There is a head line for each variant reported, followed by the alignment of supporting reads to the reference on the second line.
The breakpoints are specified after “BP”. Due to microhomology around the breakpoints, the breakpoint coordinates may shift both upstream and downstream,’BP_range’ is used to indicate the range of this shift. The header line contains the following data:
- The index of the indel/SV
- The type of indel/SV: I for insertion, D for deletion, INV for inversion, TD for tandem duplication
- The length of the SV
- “NT”:indicate that the next number is the length of non-template sequences inserted; insertions are fully covered by the NT-fields, deletions can have NT bases if the deletion is not ‘pure’, meaning that while bases have been deleted, some bases have been inserted between the breakpoints
- the length(s) of the NT fragment(s)
- the sequence(s) of the NT fragment(s) 7-8) the identifier of the chromosome the read was found on 9-10-11) BP: the start and end positions of the SV 12-13-14) BP_range: if the exact position of the SV is unclear since bases at the edge of one read-half could equally well be appended to the other read-half. In the deletion example, ACA could be on any side of the gap, so the original deletion could have been between 1337143 and 1338815, between 1337144 and 1338816, or between 1337145 and 133817, or between 1337146 and 133818. BP-range is used to indicate this range
- “Supports”: announces that the total count of reads supporting the SV follow
- The number of reads supporting the SV
- The number of unique reads supporting the SV (so not counting duplicate reads)
- +: supports from reads whose anchors are upstream of the SV 19-20) total number of supporting reads and unique number of supporting reads whose anchors are upstream of the SV
- -: supports from reads whose anchors are downstream of the SV 22-23) total number of supporting reads and unique number of supporting reads whose anchors are downstream of the SV 24-25) S1: a simple score, (“19)” + 1)* (“22)” + 1) 26-27) SUM_MS: sum of mapping qualities of anchor reads.The reads with variants or unmapped are called split-read, whose mate is called anchor reads. We use anchor reads to narrow down the search space to speed up and increase sensitivity
- the number of different samples scanned 29-30-31) NumSupSamples: the number of samples supporting the SV, as well as the number of samples having unique reads supporting the SV (in practice, these numbers are the same) 32+) Per sample: the sample name, followed by the total number of supporting reads whose anchors are upstream, the total number of unique supporting reads whose anchors are upstream, the total number of supporting reads whose anchors are downstream, and finally the total number of unique supporting reads whose anchors are downstream
The second line shows the reference.
The third line and further show the sequence of the read, whether the anchor read is upstream(+) or downstream(-), the position of the mapped half of the paired-end read, the mapping quality of the mapped read, the sample name, and the read identifier.
If you do not wish to understand the Pindel raw output format, please first convert the result to VCF and perform downstream analysis.
Use the command pindel2vcf to transfer raw output file to vcf:
cd pindel
./pindel2vcf -p /path/to/raw/output/of/pindel/_D -r ref.fa -R GRCh38 -d version.of.reference -v /path/to/store/output/vcf/deletion.vcf
If you care more about the high quality variants and want to do filtering by yourselves, you can design your own filtering conditions based on the information provided.